

First Principle Study on the Influence Mechanism of Impurity Gas O2 on the Adsorption Properties of Alloy ZrCo
-
摘要:
杂质气体在ZrCo合金表面的吸附行为对其储氢性能具有重要的影响。采用基于赝势平面波法的第一性原理对空气中的O2在ZrCo(110)表面的吸附行为进行了研究。对O2在合金表面所有稳定吸附构型进行吸附能分析以及电荷分析的结果表明:O2吸附的最稳定构型为B3(Zr—Co桥位),在该位点的吸附能为−8.124 eV。该构型的态密度和差分电荷密度的结果表明:O2在ZrCo(110)表面该位点的吸附属于强化学吸附,O—O键发生断裂。O原子与ZrCo(110)表面原子的成键本质为O原子的电子和周围表面原子的电子发生电子轨道重叠,即O原子的2s、2p轨道电子与表面的Zr原子的4p、4d轨道电子和Co原子的3d轨道电子发生了电子轨道重叠,出现了轨道杂化现象。研究结果对后续揭示ZrCo合金储氢材料在杂质气体中的毒化机制具有积极作用。
Abstract:The adsorption behavior of impurity gases on the surface of alloy ZrCo has an important influence on its hydrogen storage performance. The adsorption behavior of O2 on the ZrCo(110) surface was investigated with the first principles based on the pseudopotential plane wave method. The results of adsorption energy and charge analysis show that, the most stable geometry configuration was B3 (the Zr—Co bridge site) where the adsorption energy was –8.124 eV. The analysis of the density of states and the differential charge density show that, the adsorption behavior of O2 on the ZrCo(110) surface is a strong chemical adsorption, where the oxygen-oxygen bond breaks. The essence of bonding between atom O and the ZrCo(110) surface atom is that the electron orbit of atom O overlaps with the electron orbit of the surface atom, i.e. the 2s and 2p orbital electrons of atom O overlapped with the 4p and 4d orbital electrons of atom Zr and the 3d orbital electrons of atom Co on the surface. The research results make senses in revealing the poisoning mechanism of alloy ZrCo in impurity gases.
-
Key words:
- alloy ZrCo /
- impurity gas O2 /
- adsorption property /
- influence mechanism /
- first principle
-
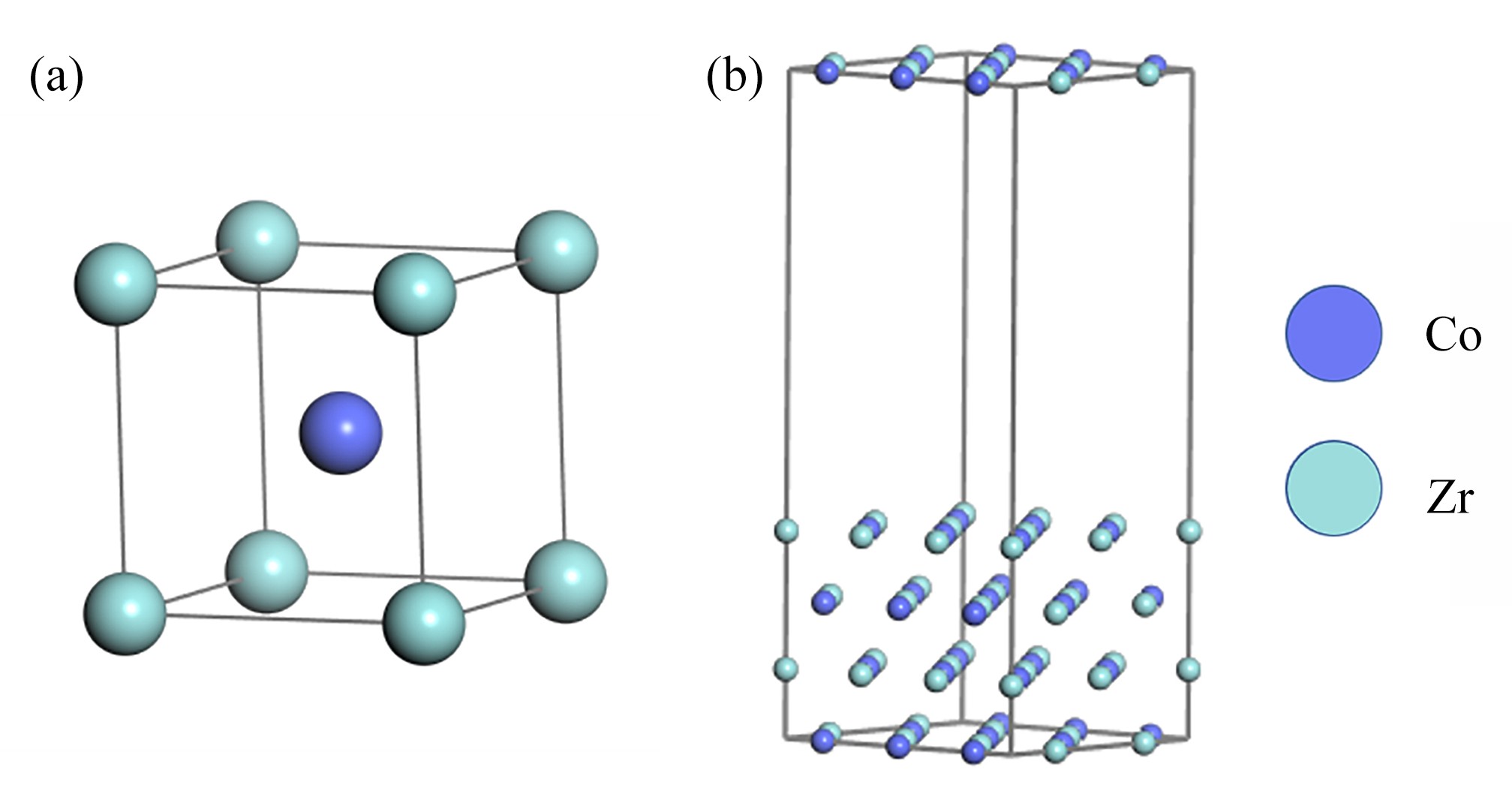
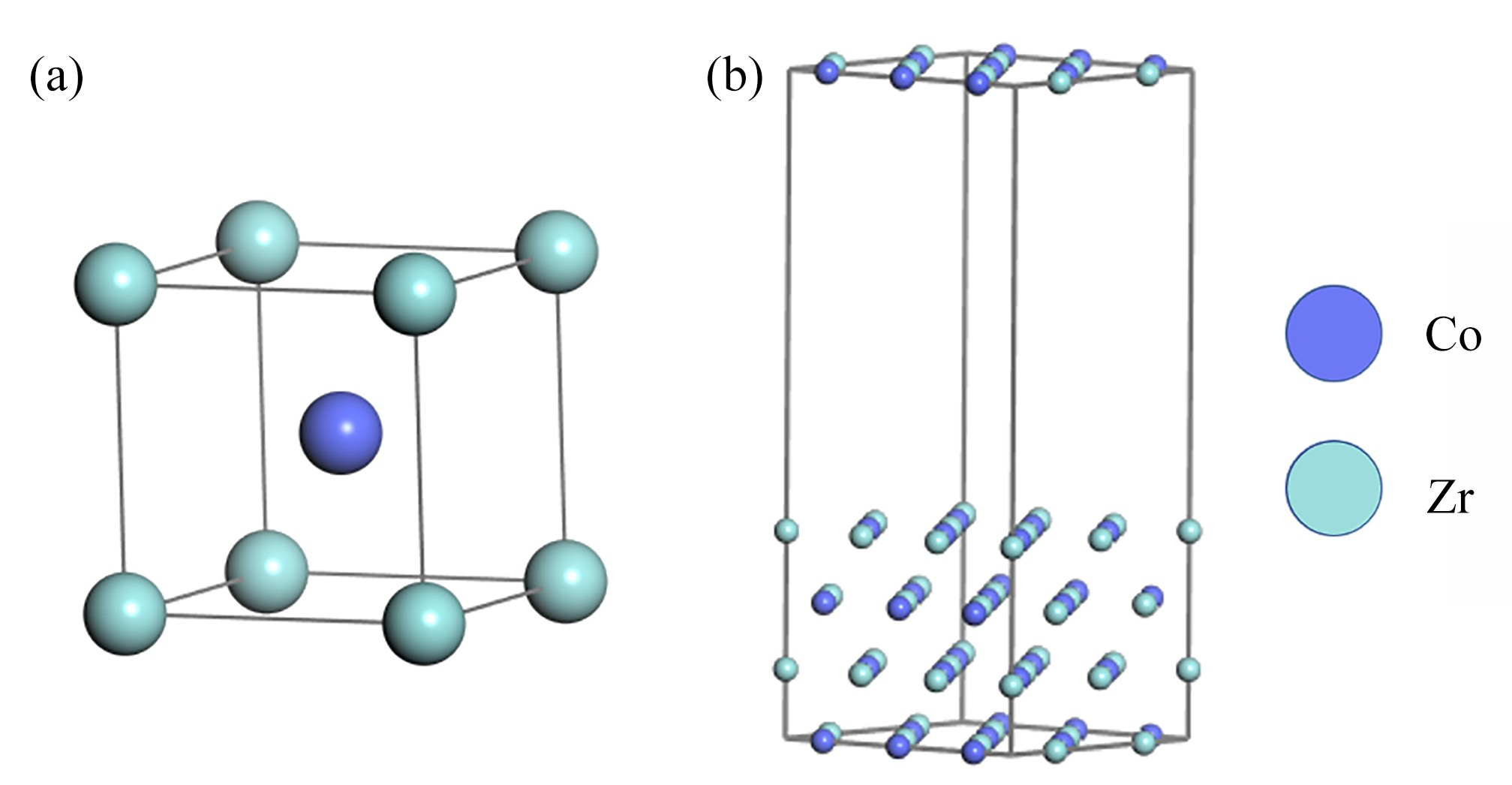
图 1 ZrCo晶胞及表面模型
注 为了解释图中的颜色,读者可以参考本文的电子网页版本,后同。
Figure 1. The unit cell structure of ZrCo and the ZrCo(110) surface model
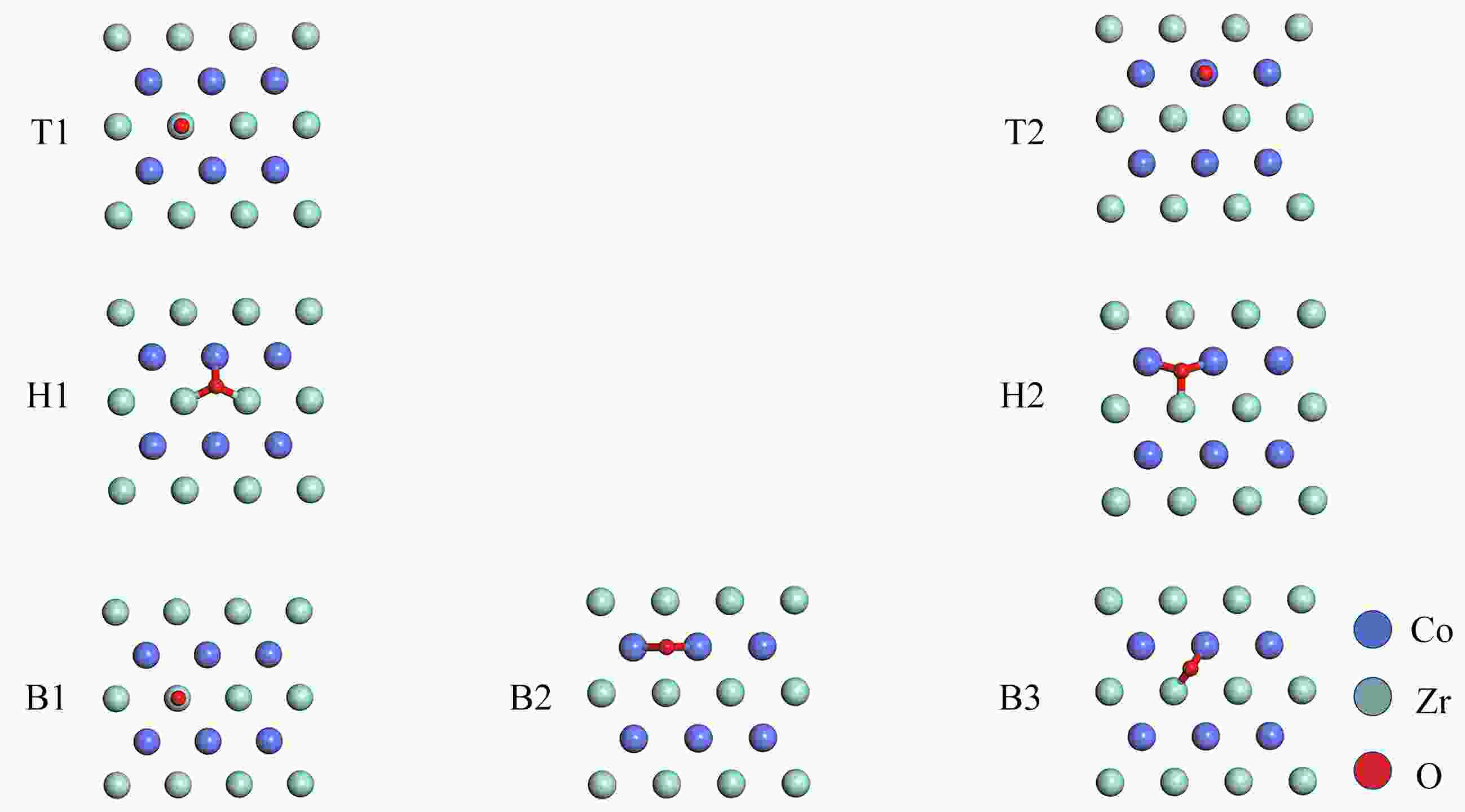
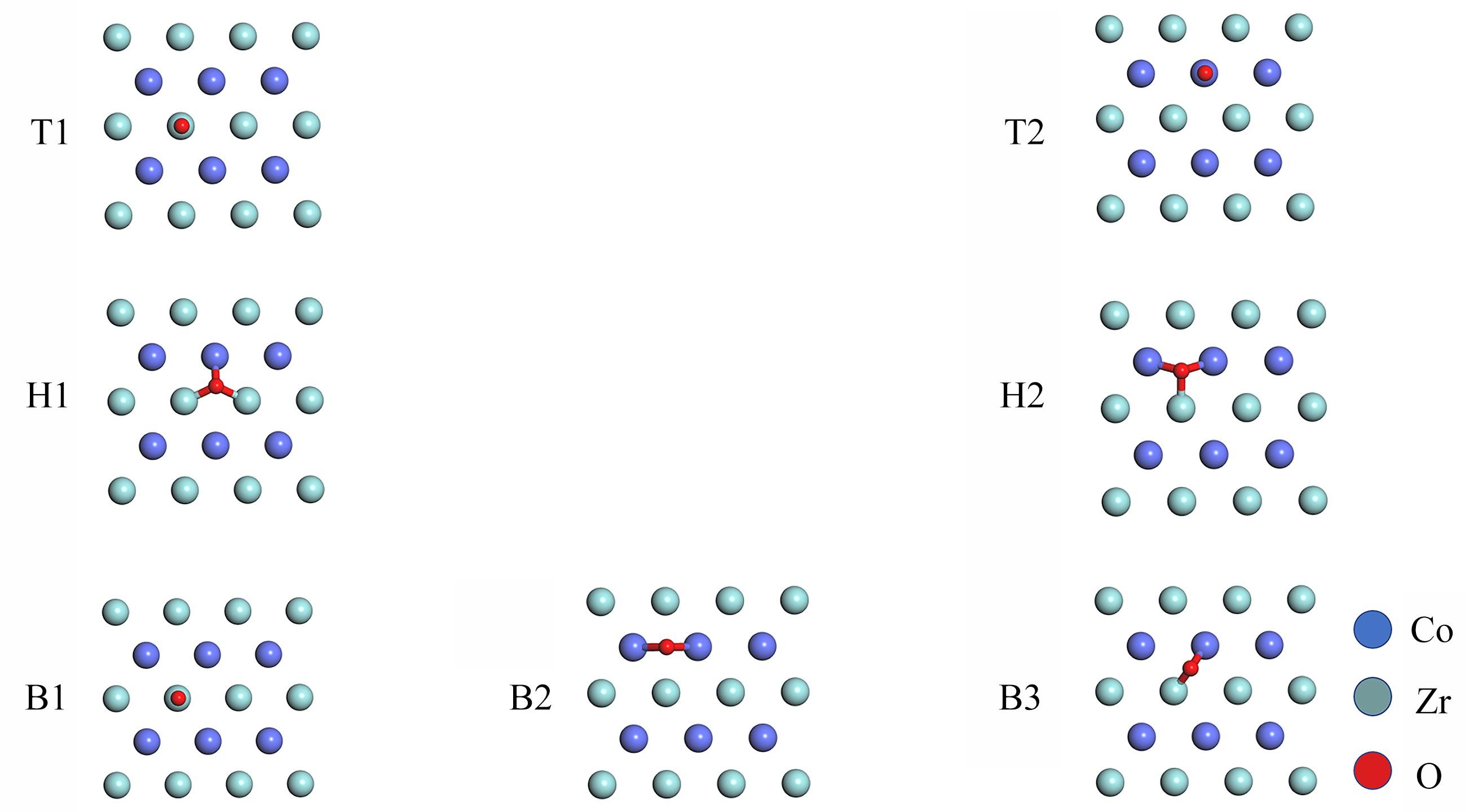
图 2 O2的所有初始吸附构型
Figure 2. All the initial configurations of O2 adsorption on the ZrCo(110) surface: top view
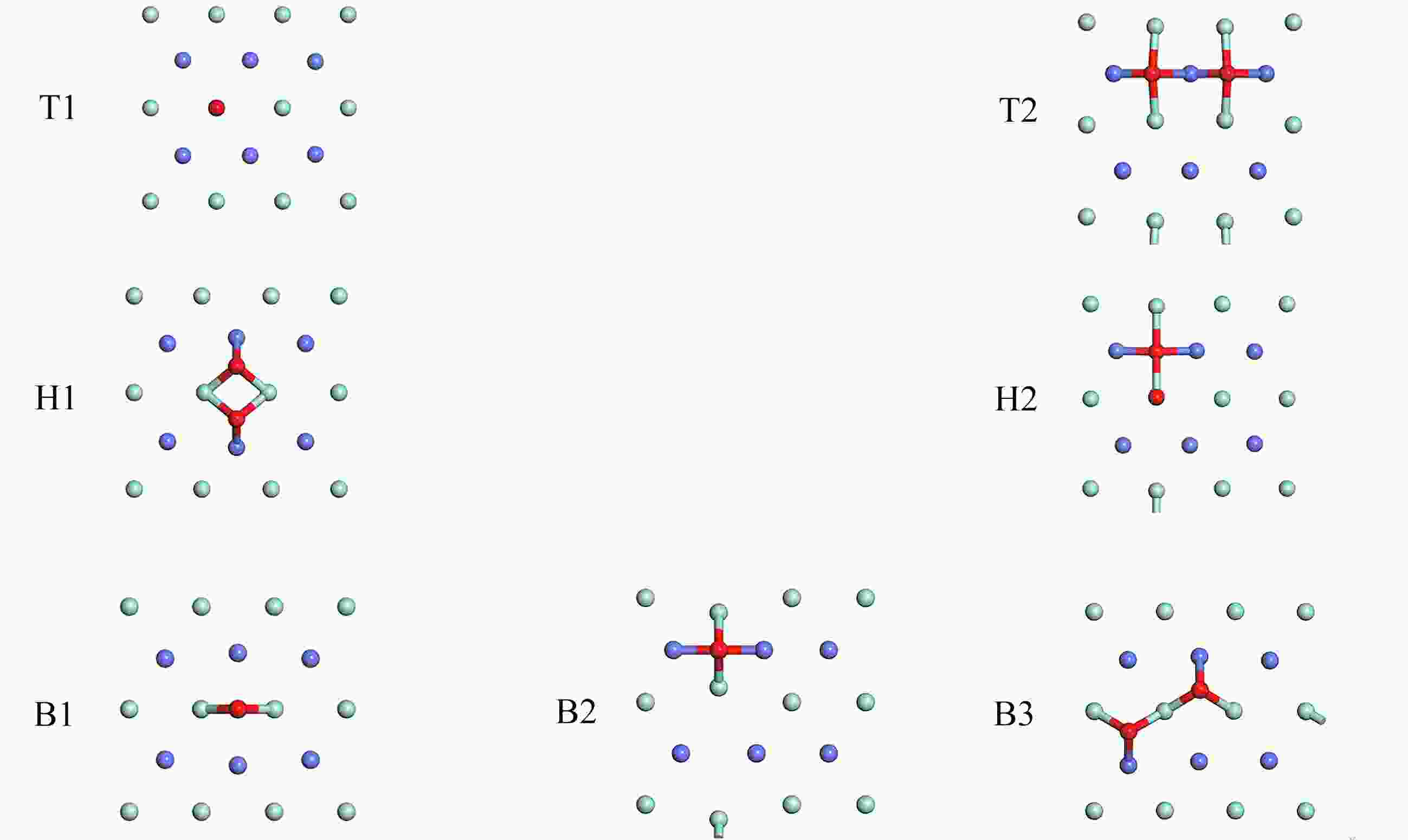
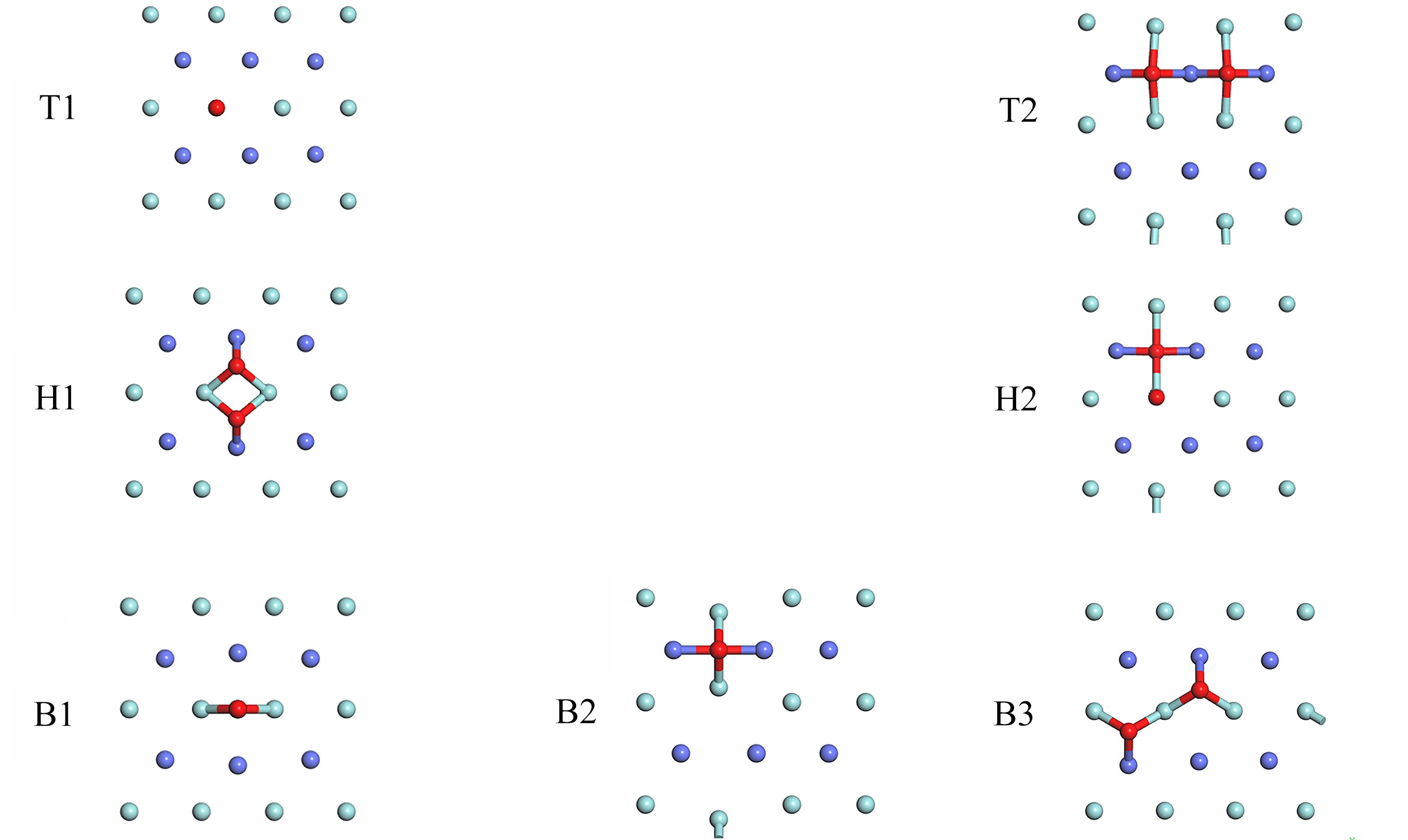
图 3 优化后O2的稳定吸附构型
Figure 3. The optimized stable adsorption of O2 on the ZrCo(110) surface
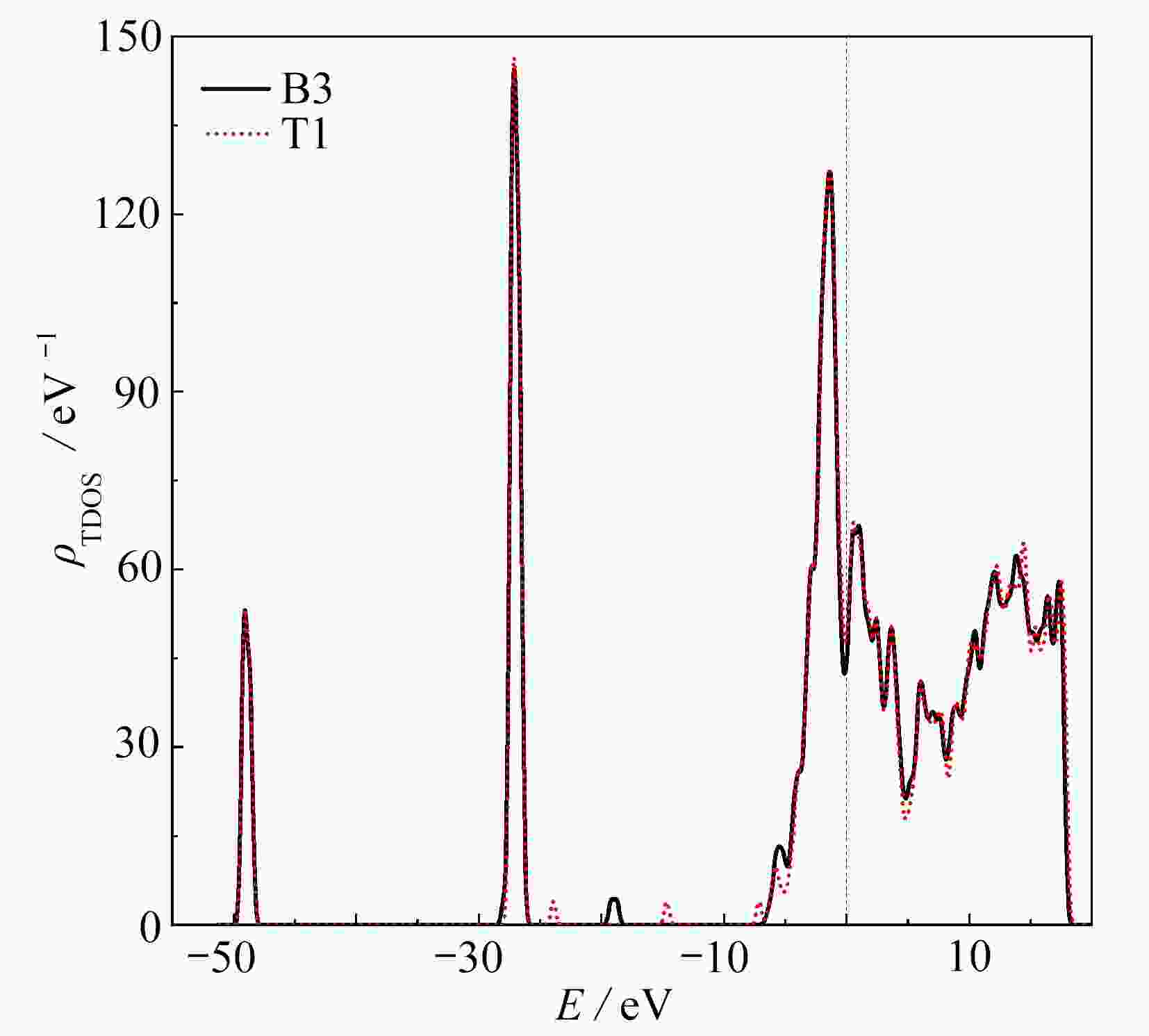
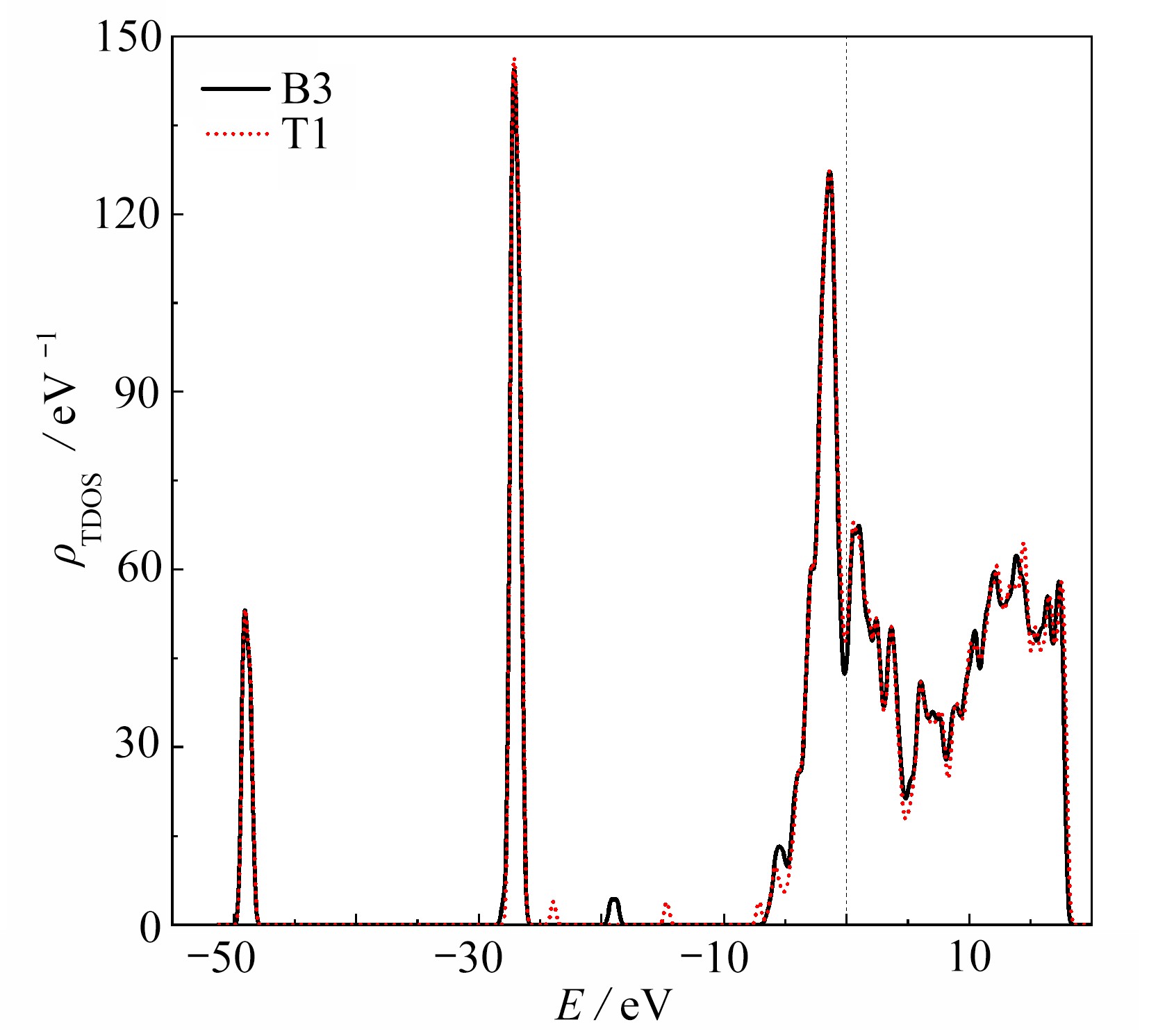
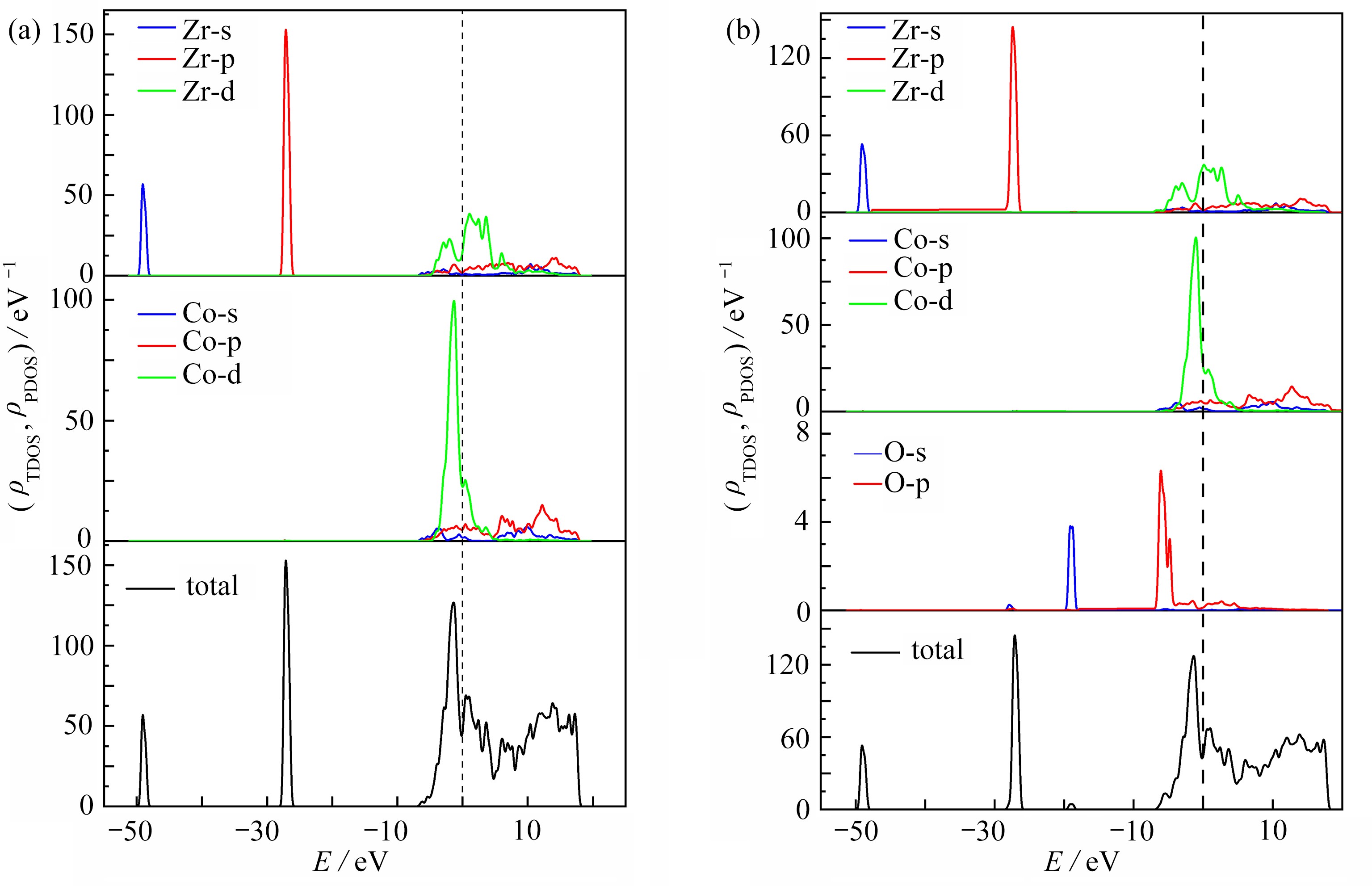
图 4 O2在ZrCo(110) B3和T1吸附位置的态密度
Figure 4. The density of states of the ZrCo(110) surface after adsorption of O2 at B3 and T1
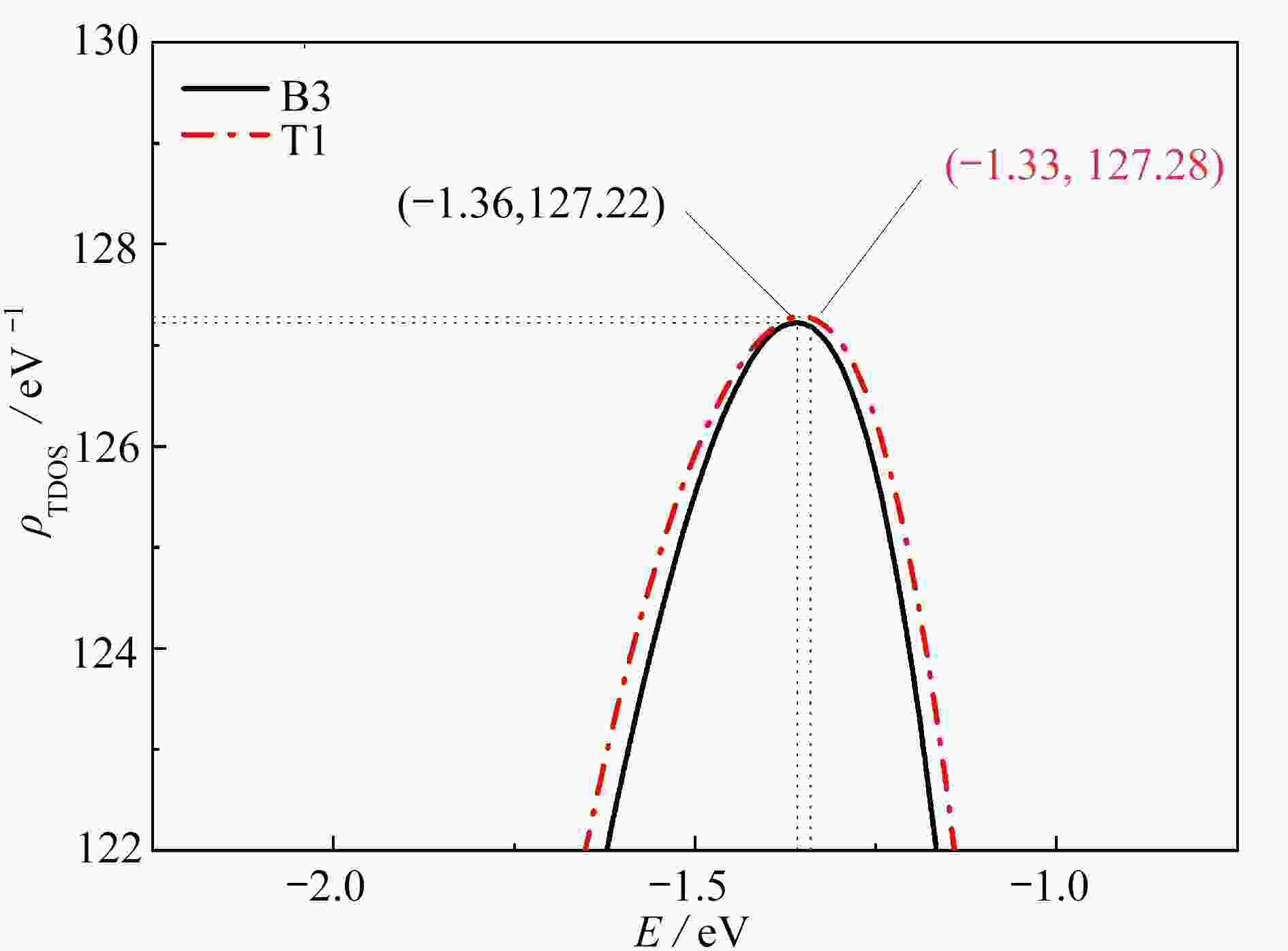
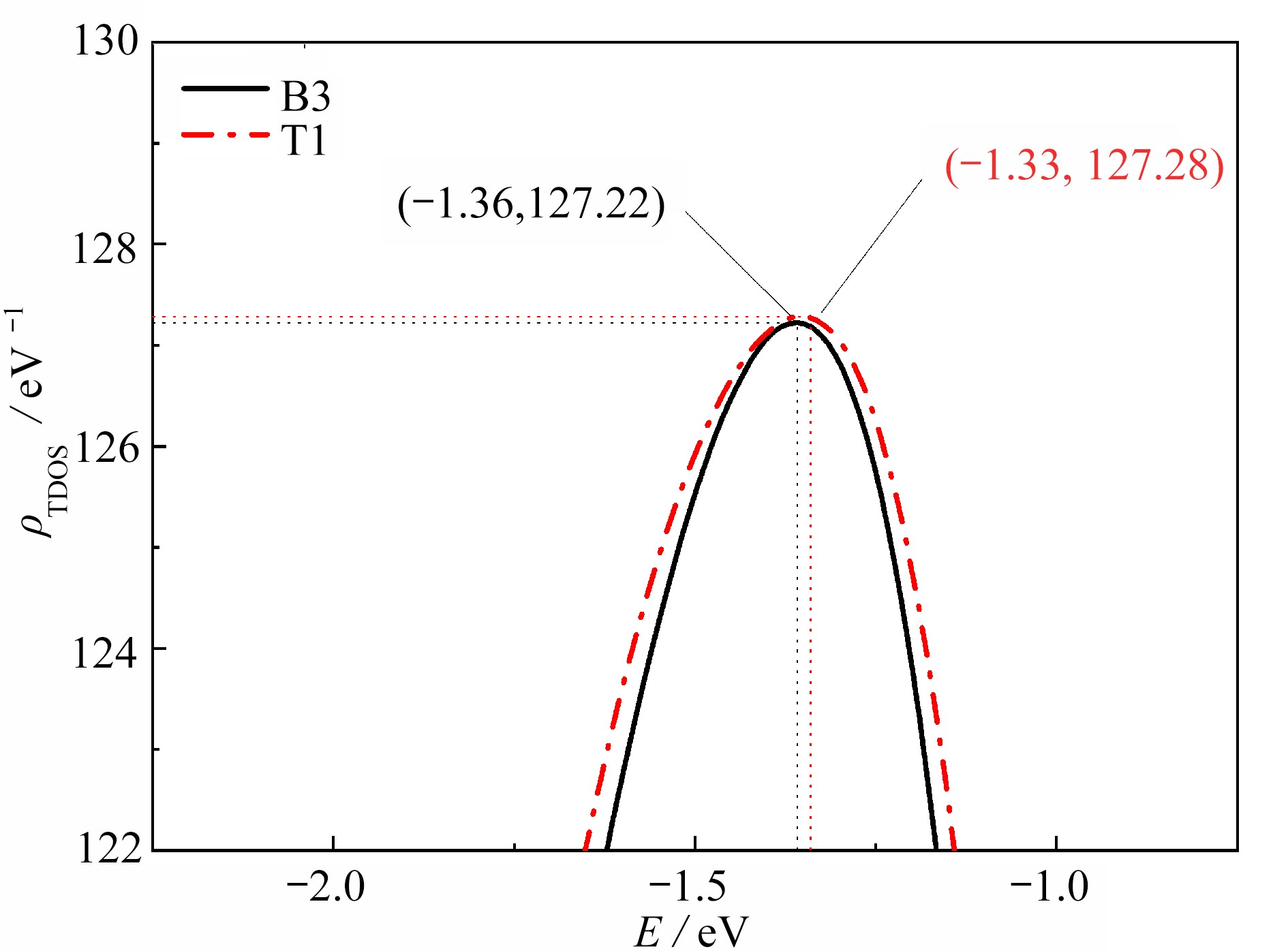
图 5 O2在ZrCo(110) B3和T1吸附后的Fermi能级附近的主态密度峰
Figure 5. Dominant DOS peaks near the Fermi level after adsorption of O2 at B3 and T1
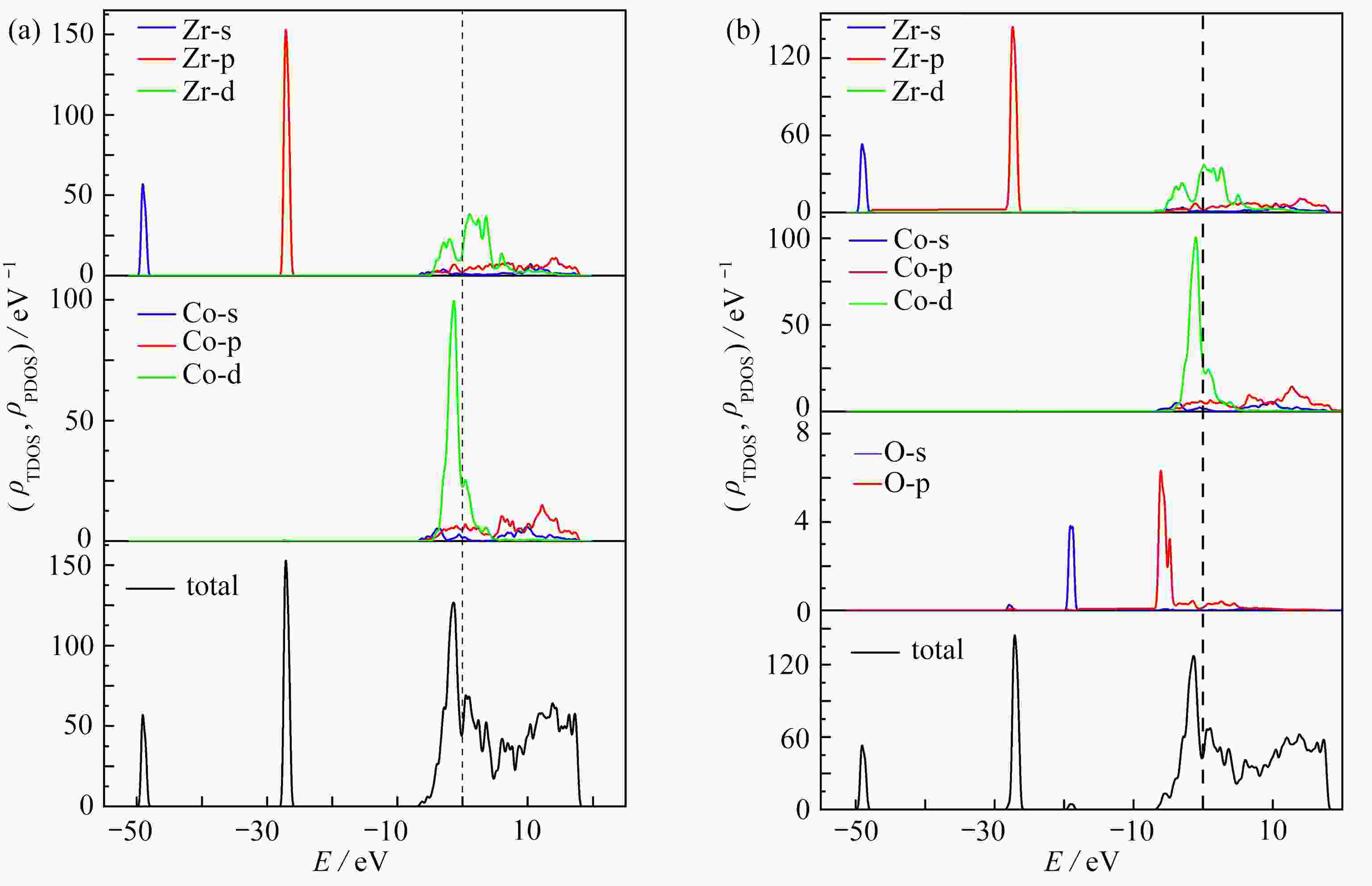
图 6 O2在ZrCo(110) B3吸附位置的态密度:(a) 吸附前;(b) 吸附后
Figure 6. The density of states of the ZrCo(110) surface of O2: (a) before adsorption; (b) after adsorption
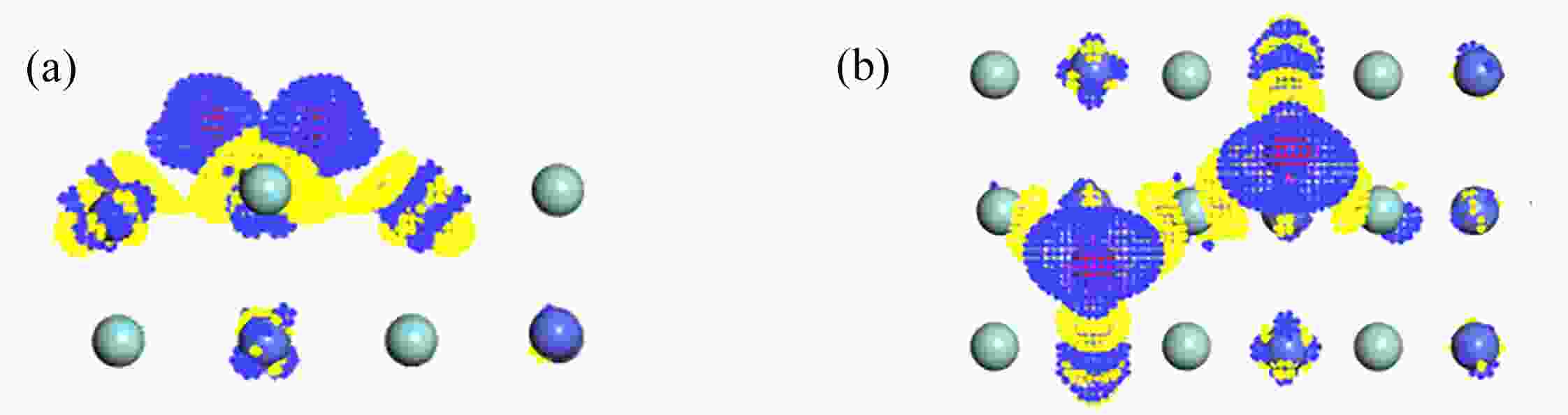
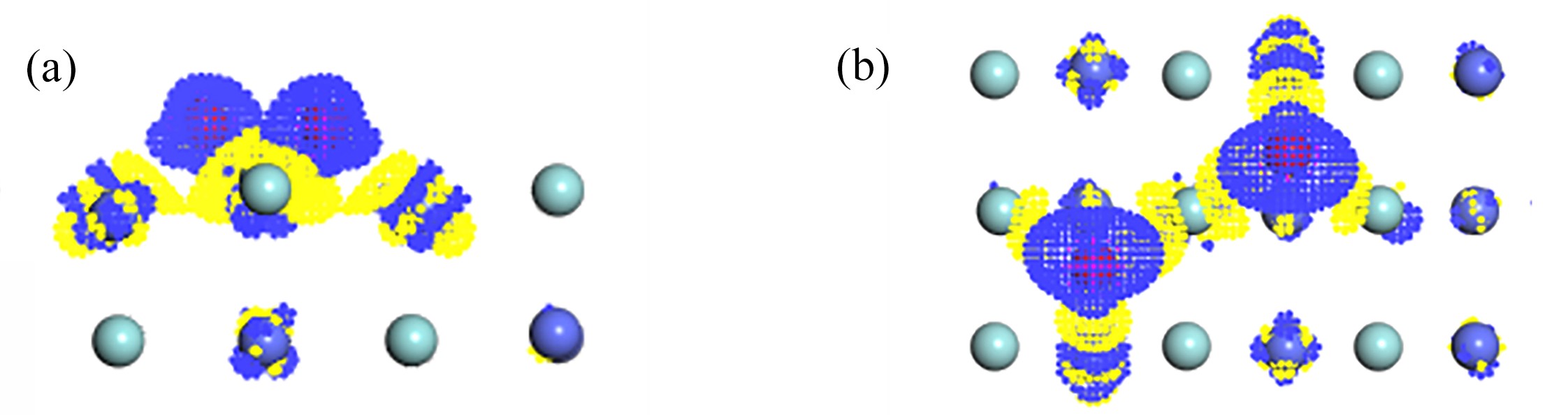
图 7 O2在B3吸附的差分电荷密度图:(a) 侧视图;(b) 俯视图
Figure 7. The differential charge density of O2 adsorption at B3 on the ZrCo(110) surface: (a) the side view; (b) the top view
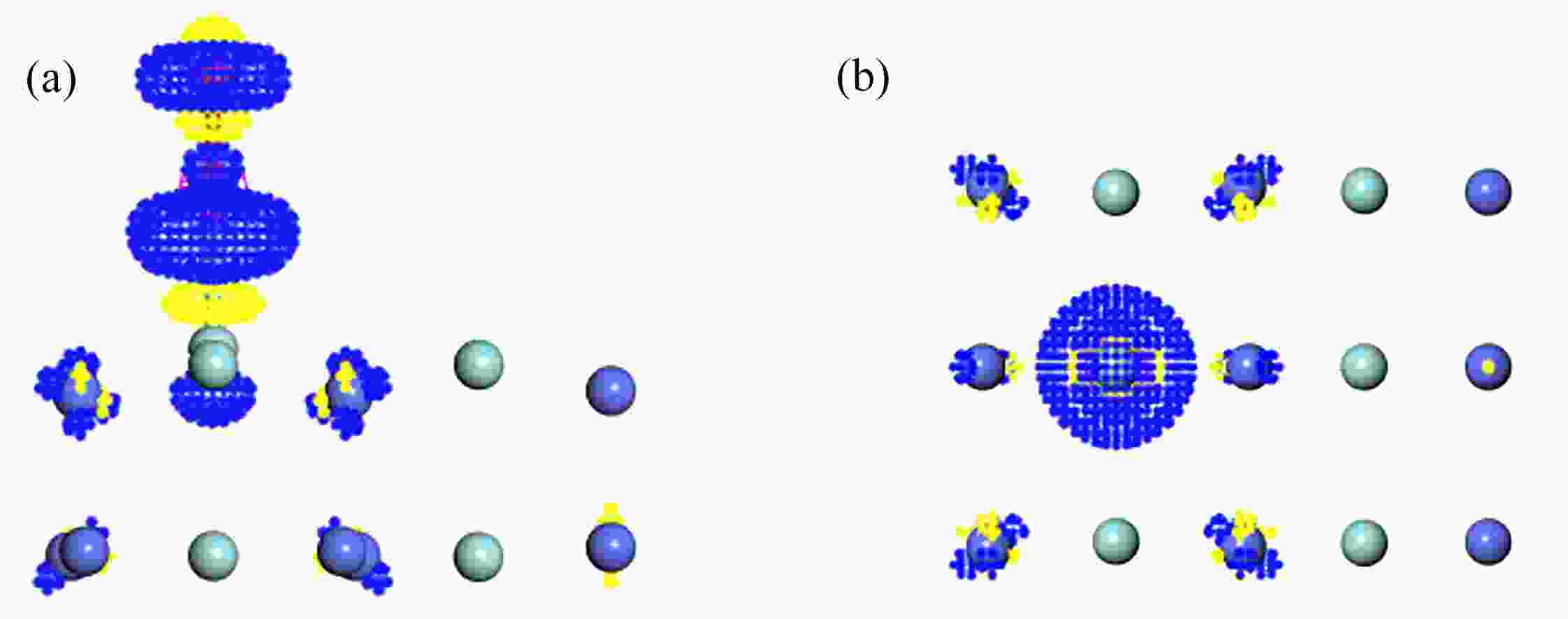
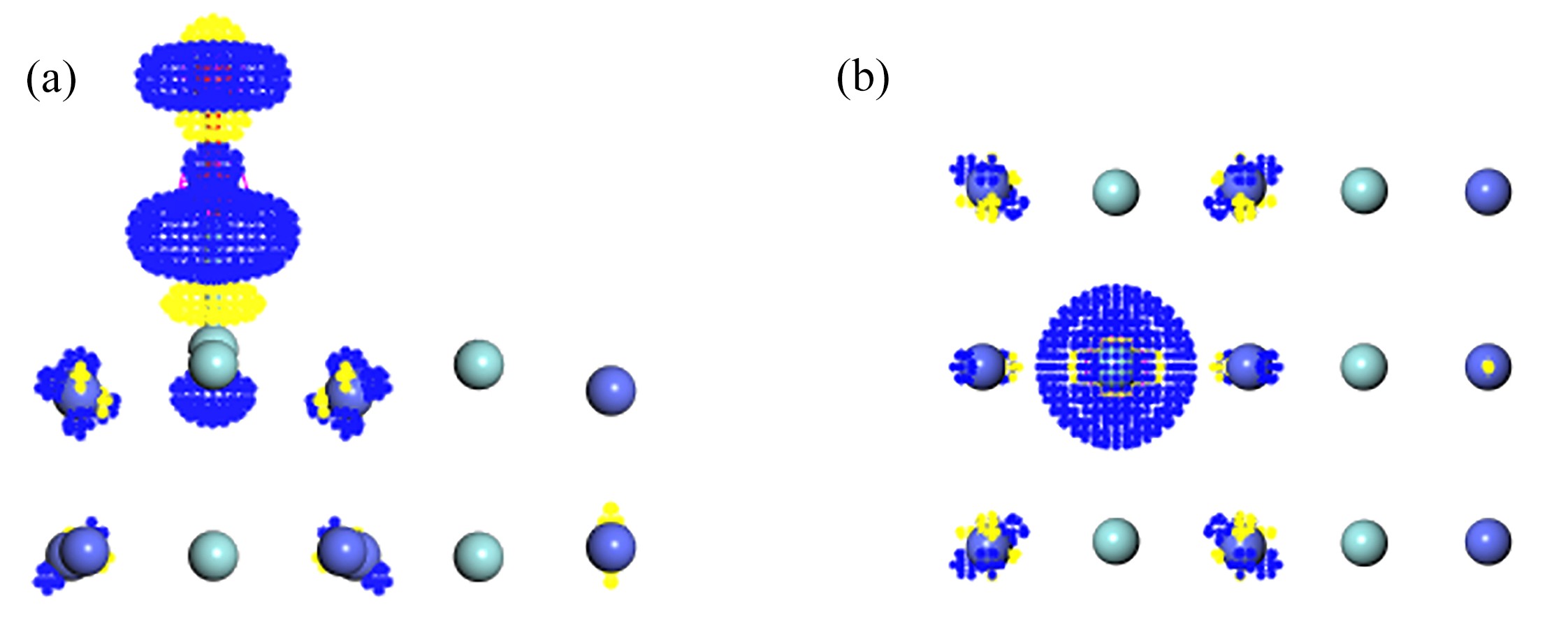
图 8 O2在T1吸附的差分电荷密度图:(a) 侧视图;(b) 俯视图
Figure 8. The differential charge density of O2 adsorption at T1 on the ZrCo(110) surface: (a) the side view; (b) the top view
表 1 ZrCo晶格常数
Table 1. Experimental and calculated lattice parameters for the ZrCo
 下载: 导出CSV
下载: 导出CSV
表 2 Zr和Co原子弛豫前后的z坐标(单位:Å)
Table 2. The z coordinates of Zr and Co atoms after relaxation for ZrCo(110) (unit: Å)
layer before relaxation after relaxation difference of z coordinate after relaxation Co (1) 6.748 6.479 −0.269 Zr (1) 6.748 6.805 0.057 Co (2) 4.498 4.603 0.105 Zr (2) 4.498 4.504 0.006
下载: 导出CSV
表 3 O2在ZrCo(110)表面稳定吸附构型的结构参数和吸附能
Table 3. The structural parameters and energy of stable adsorption configuration of O2 on the ZrCo(110) surface
model d0/Å chemical bond 1 d1/Å chemical bond 2 d2/Å Eads/eV T1 1.309 O1—Zr20 1.918 − − −0.027 T2 3.472 O1—Co16 1.879 O2—Co24 1.924 −5.786 O1—Co20 1.935 O2—Co20 1.929 O1—Zr20 2.216 O2—Zr12 2.193 O1—Zr8 2.218 O2—Zr24 2.197 H1 2.456 O1—Co20 1.835 O2—Co8 1.836 −6.782 O1—Zr20 2.100 O2—Zr20 2.099 O1—Zr24 2.100 O2—Zr24 2.099 H2 3.203 O1—Co20 1.969 O2—Zr20 1.771 −5.406 O1—Co16 1.969 − − O1—Zr8 2.206 − − O1—Zr20 2.231 − − O1—Zr19 2.238 − − B1 1.369 O1—Zr24 2.097 − − −0.513 O1—Zr20 2.098 − − B2 2.435 O1—Co20 1.958 O2—Zr20 2.028 −3.789 O1—Co16 1.959 O2—Zr8 2.030 O1—Zr20 2.024 − − O1—Zr8 2.033 − − O1—Zr19 2.042 − − B3 3.724 O1—Co20 1.952 O2—Co4 1.955 −8.124 O1—Zr20 2.041 O2—Zr16 2.039 O1—Zr24 2.046 O2—Zr20 2.046
下载: 导出CSV
表 4 O原子吸附后的净电荷分布
Table 4. The net charge distribution of O1 and O2 in each structure
model O1 O2 Qtotal T1 −0.27 −0.30 −0.57 T2 −0.68 −0.68 −1.36 H1 −0.65 −0.65 −1.3 H2 −0.70 −0.55 −1.25 B1 −0.39 −0.39 −0.78 B2 −0.63 −0.69 −1.32 B3 −0.68 −0.69 −1.37
下载: 导出CSV
-
[1] REN J W, MUSYOKA N M, LANGMI H W, et al. Current research trends and perspectives on materials-based hydrogen storage solutions: a critical review[J]. International Journal of Hydrogen Energy, 2017, 42(1): 289-311. doi: 10.1016/j.ijhydene.2016.11.195 [2] 高伟业, 张赛, 张杰, 等. 含湿相变粗糙多孔材质的热质耦合分形研究[J]. 应用数学和力学, 2022, 43(5): 561-568GAO Weiye, ZHANG Sai, ZHANG Jie, et al. Thermo-mass coupling fractal study of wet phase-change rough porous materials[J]. Applied Mathematics and Mechanics, 2022, 43(5): 561-568.(in Chinese) [3] 董彦辰, 张业伟, 陈立群. 惯容器非线性减振与能量采集一体化模型动力学分析[J]. 应用数学和力学, 2019, 40(9): 968-979DONG Yanchen, ZHANG Yewei, CHEN Liqun. Dynamic analysis of the nonlinear vibration absorber-energy harvester integration model with inerters[J]. Applied Mathematics and Mechanics, 2019, 40(9): 968-979.(in Chinese) [4] ZHANG F, ZHAO P C, NIU M, et al. The survey of key technologies in hydrogen energy storage[J]. International Journal of Hydrogen Energy, 2016, 41(33): 14535-14552. doi: 10.1016/j.ijhydene.2016.05.293 [5] RAJAURA R S, SRIVASTAVA S, SHARMA P K, et al. Structural and surface modification of carbon nanotubes for enhanced hydrogen storage density[J]. Nano-Structures & Nano-Objects, 2018, 14: 57-65. [6] KOJIMA Y. Hydrogen storage materials for hydrogen and energy carriers[J]. International Journal of Hydrogen Energy, 2019, 44(33): 18179-18192. doi: 10.1016/j.ijhydene.2019.05.119 [7] WANG F, LI R F, DING C P, et al. Recent progress on the hydrogen storage properties of ZrCo-based alloys applied in international thermonuclear experimental reactor (ITER)[J]. Progress in Natural Science: Materials International, 2017, 27(1): 58-65. doi: 10.1016/j.pnsc.2016.12.018 [8] LIANG Z Q, XIAO X Z, YAO Z D, et al. A new strategy for remarkably improving anti-disproportionation performance and cycling stabilities of ZrCo-based hydrogen isotope storage alloys by Cu substitution and controlling cutoff desorption pressure[J]. International Journal of Hydrogen Energy, 2019, 44(52): 28242-28251. doi: 10.1016/j.ijhydene.2019.09.077 [9] YAO Z D, XIAO X Z, LIANG Z Q, et al. Improvement on the kinetic and thermodynamic characteristics of Zr1–xNbxCo (x = 0~0.2) alloys for hydrogen isotope storage and delivery[J]. Journal of Alloys and Compounds, 2019, 784: 1062-1070. doi: 10.1016/j.jallcom.2019.01.100 [10] KOU H Q, SANG G, LUO W H, et al. Comparative study of full-scale thin double-layered annulus beds loaded with ZrCo, Zr0.8Hf0.2Co and Zr0.8Ti0.2Co for recovery and delivery of hydrogen isotopes[J]. International Journal of Hydrogen Energy, 2015, 40(34): 10923-10933. doi: 10.1016/j.ijhydene.2015.06.153 [11] ZHAO Y M, LI R F, TANG R H, et al. Effect of Ti substitution on hydrogen storage properties of Zr1−xTixCo (x = 0, 0.1, 0.2, 0.3) alloys[J]. Journal of Energy Chemistry, 2014, 23(1): 9-14. doi: 10.1016/S2095-4956(14)60111-X [12] WENG C C, XIAO X Z, HUANG X, et al. Effect of Mn substitution for Co on the structural, kinetic, and thermodynamic characteristics of ZrCo1–xMnx (x = 0~0.1) alloys for tritium storage[J]. International Journal of Hydrogen Energy, 2017, 42(47): 28498-28506. doi: 10.1016/j.ijhydene.2017.09.157 [13] GLUGLA M, LÄSSER R, DÖRR L, et al. The inner deuterium/tritium fuel cycle of ITER[J]. Fusion Energy and Design, 2003, 69: 39-43. doi: 10.1016/S0920-3796(03)00231-X [14] ZHANG G H, TANG T, SANG G, et al. Effect of Ti modification on hydrogenation properties of ZrCo in the presence of CO contaminant gas[J]. Rare Metal Materials and Engineering, 2017, 46(11): 3366-3373. [15] GARRITY K F, BENNETT J W, RABE K M, et al. Pseudopotentials for high-throughput DFT calculations[J]. Computational Materials Science, 2014, 81: 446-452. doi: 10.1016/j.commatsci.2013.08.053 [16] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868. doi: 10.1103/PhysRevLett.77.3865 [17] CHATTARAJ D, PARIDA S C, DASH S, et al. Structural, electronic and thermodynamic properties of ZrCo and ZrCoH3: a first-principles study[J]. International Journal of Hydrogen Energy, 2012, 37(24): 18952-18958. doi: 10.1016/j.ijhydene.2012.09.108 [18] GACHON J C, SELHAOUI N, ABA B, et al. Comparison between measured and predicted enthalpies of formation[J]. Journal of Phase Equilibria, 1992, 13: 506-511. doi: 10.1007/BF02665763 [19] WANG Q Q, KONG X G, YU Y, et al. Influence of the Fe-doping on hydrogen behavior on the ZrCo surface[J]. International Journal of Hydrogen Energy, 2021, 46(68): 33877-33888. doi: 10.1016/j.ijhydene.2021.07.198 [20] 蒙大桥, 罗文华, 李赣, 等. Pu(100)表面吸附CO2的密度泛函研究[J]. 物理学报, 2009, 58(12): 8224-8229 doi: 10.3321/j.issn:1000-3290.2009.12.017MENG Daqiao, LUO Wenhua, LI Gan, et al. Density functional study of CO2 adsorption on Pu(100) surface[J]. Acta Physica Sinica, 2009, 58(12): 8224-8229.(in Chinese) doi: 10.3321/j.issn:1000-3290.2009.12.017 [21] DEVILLERS M, SIRCH M, PENZHORN R D. Hydrogen-induced disproportionation of the intermetallic zirconium-cobalt compound ZrCo[J]. Chemistry of Materials, 1992, 4(3): 631-639. doi: 10.1021/cm00021a025 [22] CHEN Q, HUANG Z W, ZHAO Z D, et al. Thermal stabilities, elastic properties and electronic structures of B2-MgRe (Re=Sc, Y, La) by first-principles calculations[J]. Computational Materials Science, 2013, 67: 196-202. doi: 10.1016/j.commatsci.2012.08.010 [23] WANG L S, DING J, HUANG X, et al. Influence of Ti/Hf doping on hydrogen storage performance and mechanical properties of ZrCo compounds: a first principle study[J]. International Journal of Hydrogen Energy, 2018, 43(29): 13328-13338. doi: 10.1016/j.ijhydene.2018.05.061 -
计量
- 文章访问数: 517
- HTML全文浏览量: 195
- PDF下载量: 43
- 被引次数: 0

 渝公网安备50010802005915号
渝公网安备50010802005915号